
Email:
sales@chinesepeptide.com
Feb 08, 2024


Antimicrobial Peptides (AMPs) play a crucial role in the innate and acquired immune responses to pathogenic attacks and are present in almost every living thing, including bacteria, fungi, plants, invertebrates, and vertebrates. Immune cells (granulocytes and macrophages), epithelial cells (vaginal epithelium, lung epithelium, and oral cavity epithelium), and the small intestine are only a few of the cells and organelles that release AMPs. Zeya and Spitznagel reported the antibacterial properties of peptides for the first time in 1966.[1-4] They discovered several of the peptides contained basic amino acids like arginine and lysine when studying lysosomal cationic proteins and peptides. They quickly used experiments to show that certain gram-positive and gram-negative bacteria were resistant to the antibacterial activity of lysosomal cationic proteins (LCPs).

Figure 1. Representative models of common antimicrobial peptides (AMPs) showing alpha-helical, beta-sheet, amphiphilic structural motifs that relate form and function. Molecular graphics images were produced using the Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081).[17]
The growth of multi-drug-resistant bacteria due to antibiotic misuse is one of the main drivers for the development of peptide antibiotics. In the U.S. and Europe alone, antimicrobial-resistant diseases claimed over 50,000 deaths in 2013, according to the European Centre for Disease Prevention and Control Antimicrobial Resistance Interactive Database (EARS-NET).[5] For instance, 10% of blood infections caused by Staphylococcus aureus are caused by methicillin-resistant strains in numerous European nations.[6] According to reports, these methicillin-resistant bacterial strains are present in much higher amounts in some nations. Small-molecule antibiotic producers have attempted to address this issue by altering the structures of currently available antibiotics and creating new antibiotic classes. Despite some of these valiant attempts, antibiotic resistance has become a serious global health concern.
Alpha-helices, beta-sheets, loops, and extended structures are only a few of the structural motifs that are known to adopt secondary forms in AMPs (Figure 1).[7-9] A variety of antimicrobial activities, including membrane disruptors, effector molecules, immunomodulators, intracellular proteins, and human contraceptives, are made possible by the wide range of structural motifs and characteristics.[15,16] The majority of AMPs have polycationic charges between +2 and +9, which allows them to electrostatically attach the negatively charged bacterial membrane surface. Cationic AMPs bind the anionic phosphate groups in the outer membrane of gram-negative bacteria. On the other hand, gram-positive bacteria don't have an outer membrane or lipopolysaccharides on their surface to bind AMPs. Instead, the peptidoglycans on the surface of these bacteria emit anionic teichoic acid to draw AMPs. The ability of AMPs to discriminate between bacteria and host cells is provided by the lipopolysaccharides (LPS) and peptidoglycans present on the surface of bacteria. AMPs are far less harmful than conventional small molecule antibiotics due to their capacity to distinguish between host and bacterial cells.
Alpha-helices, beta-sheets, loops, and extended structures are only a few of the structural motifs that are known to adopt secondary forms in AMPs (Figure 1).[7-9] A variety of antimicrobial activities, including membrane disruptors, effector molecules, immunomodulators, intracellular proteins, and human contraceptives, are made possible by the wide range of structural motifs and characteristics.[15,16] The majority of AMPs have polycationic charges between +2 and +9, which allows them to electrostatically attach the negatively charged bacterial membrane surface. Cationic AMPs bind the anionic phosphate groups in the outer membrane of gram-negative bacteria. On the other hand, gram-positive bacteria don't have an outer membrane or lipopolysaccharides on their surface to bind AMPs. Instead, the peptidoglycans on the surface of these bacteria emit anionic teichoic acid to draw AMPs. The ability of AMPs to discriminate between bacteria and host cells is provided by the lipopolysaccharides (LPS) and peptidoglycans present on the surface of bacteria. AMPs are far less harmful than conventional small-molecule antibiotics due to their capacity to distinguish between host and bacterial cells. The charge distribution, shape, and secondary structures of the AMP determine how it interacts with the membranes. Studies comparing the CD spectra of different AMPs in the presence of liposomes demonstrate that some AMPs are capable of changing their conformation in hydrophobic environments. For instance, melittin exhibits random coil behavior in solution but adopts a -helical shape when it comes into contact with membrane surfaces (Table 1). Many other AMPs, such as defensins, which include numerous disulfide bridges, have stable -sheet structures that are crucial to their modes of action. AMPs target intracellular proteins and membranes, such as the endoplasmic reticulum membrane, to cause apoptosis or necrosis, or they target bacterial cellular membranes to cause membrane lysis or disruption.
Electrostatic interactions between the membrane and the AMP are the first step in the insertion or disruption of bacterial cell membranes by AMPs. Following this electrostatic pairing interaction, AMP is absorbed in the outer membrane of the bacterial cell (in the case of gram-negative bacteria), where hydrogen bonds are formed between the basic amino acid residues in the peptide (such as lysine and arginine) and the phosphate groups exposed on the membrane surface. Initiating the process of membrane instability, this action results in the dissolution of salt bridges between the phosphate groups and nearby cations. The close packing structure of the membrane is further disrupted by hydrophobic interactions between uncharged residues (leucine, isoleucine, valine, tryptophan, and phenylalanine) and hydrophobic carbon chains of Lipopolysaccharides (LPS), which further causes disorganization. When a certain level of AMPs is achieved surrounding the bacterium, a process known as "membrane thinning" occurs in which the membrane thins, leading to lateral expansions and an increase in water translocation across the membrane. The membrane is ruptured, the membrane potentials are decreased, and cell death results.
Pore formation causes intracellular components to be promptly evacuated, as opposed to membrane thinning, which results in fast cell death. The barrel-stave or toroidal model can be used to describe how the pores form, which is influenced by the AMP's structure. The hydrophobic portions of the peptide (i.e., hydrophobic amino acid residues) associate and align with the hydrocarbon core of the lipids through a process known as "hydrophobic matching" in the barrel-stave model, where AMPs aggregate and subsequently enter into the membrane bilayer. The inside surface of the pore is formed by the hydrophilic areas of the AMP, allowing hydrophilic molecules of the right size to pass through effortlessly. AMPs have fewer hydrophobic interactions and can enter the membrane more deeply in the toroidal pore model than in the barrel-stave model. The polar head groups in the lipid bilayer are drawn into the bilayer by AMPs due to electrostatic interactions, which results in membrane curvature and packing disruption. Good examples of AMPs that cause toroidal holes include LL-37, melittin, and magainin; the latter forms pores with a diameter of 2 to 3 nm that only permit water-sized molecules to escape. Pore diameters are likewise concentration-dependent; for melittin, only temporary pores form at low concentrations, whereas significantly bigger (3+ nm) more persistent pores form at high AMP concentrations.
| PEPTIDE | SEQUENCE | STRUCTURE | MECHANISM | REFERENCES |
| Histatin 5 | DSHAKRHHGYKRKFHEKHHSHRGY | Histidine-Rich | ROS generation | M.Nishikata et al., Biochem. Biophys. Res. Commun., 174, 625 (1991) |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | α-Helix | Toroidal pore, carpet | (a) G.H. Gudmundsson et al., Eur. J. Biochem., 238, 325 (1996); (b) Madera, Laurence, and Robert EW Hancock. Journal of Innate Immunity 4.5-6 (2012): 553-568; (c) Choi, Ka Yee G., Scott Napper, and Neeloffer Mookherjee. Immunology 143.1 (2014): 68-80. |
| Cecropin B | KWKVFKKIEKMGRNIRNGIVKAGPAIAVLGEAKAL-NH2 | α-Helix | Channel formation | D. Andreau et al., PNAS, 80, 6475 (1983) |
| Cecropin A, porcine | KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK-NH2 | α-Helix | Channel formation | D. Andreu et al., Proc. 20th Euro. Pept. Symp., 361 (1988) |
| Cecropin P1, porcine | SWLSKTAKKLENSAKKRISEGIAIAIQGGPR | α-Helix | Channel formation | J.Y. Lee et al., PNAS, 86, 9159 (1989) |
| Indolicidin | ILPWKWPWWPWRR-NH2 | Tryptophan-rich, extended | Pore | C. Subbalakshmi et al., FEBS Letters, 395, 48 (1996) |
| Defensin-1 (human) HNP-1 | ACYCRIPACIAGERRYGTCIYQGRLWAFCC (3 disulfide bridges) | β-Sheet | Pore, carpet | M.E. Selsted et al., J. Clin. Invest., 76, 1436 (1985) |
| beta-Defensin-2, human | GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP (3 disulfide bridges) | β-Sheet | Pore, carpet | Schröder, Jens-M., and Jürgen Harder. The International Journal of Biochemistry & Cell Biology 31.6 (1999): 645-651. |
| alpha-Defensin 6 | DCYCRIPACIAGERRYGTCIYQGRLWAFCC (3 disulfide bridges) | β-Sheet | Pore, carpet | P.A.Raj et al., Biochem. J., 347, 633 (2000) |
| Defensin HNP-3 (human) | DCYCRIPACIAGERRYGTCIYQGRLWAFCC (3 disulfide bridges) | β-Sheet | Pore, carpet | P.A.Raj et al., Biochem. J., 347, 633 (2000) |
| Defensin (human) HNP-2 | CYCRIPACIAGERRYGTCIYQGRLWAFCC (3 disulfide bridges) | β-Sheet | Pore, carpet | Selsted, M. and S. Harwig, J. Biol. Chem. 264, 4003 (1989) |
| beta-Defensin-1, human | DHYNCVSSGGQCLYSACPIFTKIQGTCYRGKAKCCK (3 disulfide bridges) | β-Sheet, α-helix | Pore, carpet | Hoover, David M., Oleg Chertov, and Jacek Lubkowski. Journal of Biological Chemistry 276.42 (2001): 39021-39026. |
| beta-Defensin-4, human | ELDRICGYGTARCRKKCRSQEYRIGRCPNTYACCLRK (3 disulfide bridges) | β-Sheet, α-helix | Pore, carpet | García, José-Ramón Conejo, et al. The FASEB Journal 15.10 (2001): 1819-1821. |
| beta-Defensin-3, human | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK (3 disulfide bridges) | β-Sheet, α-helix | Pore, carpet | (a) J. Harder, J. Bartels, E. Christophers, and J.-M. Schröder, J. Biol. Chem., 276, 5707 (2001) (Original); (b) L.A. Duits, et al., Biochem. Biophys. Res. Commun., 280, 522 (2001); (c) H.P. Jia, et al., Gene, 263, 211 (2001) |
| Magainin-2 | GIGKFLHSAKKFGKAFVGEIMNS | α-helix in membrane | Toroidal pore | A. Mor et al., Biochemistry, 30, 8824 (1991) |
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ-NH2 | α-helix | Toroidal pore/carpet, ROS generation | Raynor et al., J. Biol. Chem., 266, 2753 (1991) Schweitz, Toxicon, 22, 308 (1984) |
The capacity of the AMP to initially assume an orientation parallel to the membrane and then change that orientation perpendicularly to 'drill' into the lipid bilayer is a key factor in the development of membrane holes. Not all AMPs change phases; some of them continue to be parallel to the membrane. The "Carpet Model" is a name that has been given to this permeabilization process. The ability of the AMP to adopt a particular structure is not necessary to permeabilize membranes according to this hypothesis, which places less emphasis on form and secondary structures. Instead, AMPs group together at the bilayer's surface and bind to the surface of the cell while projecting their hydrophilic side beyond the cell. As AMPs build up on the outer membrane, the membrane finally breaks down due to a charge imbalance and increasing surface tension. Contrary to pore model simulations, the catastrophic breakdown of the bacterial membrane causes the ejection of all cytoplasmic contents, including big macromolecules. In order to distinguish between pore and carpet models, one might look at the size of the molecules released. The carpet model varies from the pore models in that it needs AMP concentrations that are high enough to cover the entire bacterial cell in order to function effectively. AMPs that use one of the pore processes have lower concentration requirements, making them more desirable therapeutic targets. Pore-forming AMPs, such as LL-37, melittin, magainins, and defensins, have drawn a lot of attention for therapeutic development.
AMPs kill bacteria in a variety of ways in addition to membrane disruption and puncturing. Many AMPs target intracellular proteins or membranes rather than only serving as membrane detergents by translocating across cellular membranes without rupturing them. AMPs that traverse the cell membrane, such indolicidin, HNP-1, dermaseptin, and others, stop some vital cellular processes that result in cell death. When indolicidin, a 13 amino acid peptide rich in tryptophan residues, enters a cell's cytoplasm, it forms a calcium-dependent bond with the calmodulin protein. Indolicidin thus, at nanomolar doses, inhibits calmodulin-stimulated phosphodiesterase activity.[18] Similar to Histatin 5, other AMPs also translocate into cells but attach to distinct intracellular targets.[19] The likely target of histatin toxicity is the mitochondria, essential cellular organelles involved in ATP generation and oxygen consumption. Reactive oxygen species (ROS) can be produced when mitochondrial respiration is interfered with, which can cause cell death or apoptosis even if the particular route of histatin toxicity is unknown.
Many AMPs have dual processes that involve both membrane rupture and apoptosis, although some AMPs don't seem to start acting until they enter the cell. An analog of defensin called coprisin, for instance, has broad-spectrum antifungal activity without damaging human erythrocytes.[20] It was evident that membrane disruption was not taking place after completing a number of different membrane studies, including calcium leakage measurements, 1,6-diphenyl-1,3,5-hexatriene (DPH) fluorescence assays, and rhodamine-conjugated single giant unilamellar vesicle (GUV) analysis. Reactive oxygen species (hydroxyl radicals in the case of coprisin) aid in apoptosis, just like in the case of histatin. Coprisin induces cytochrome c release and suppresses mitochondrial activity in addition to apoptosis. Coprisin can still target membrane LPS with precision while not harming the membrane.
Although AMPs were initially studied for their antimicrobial activity and their direct ability to kill bacteria, their mechanisms of action are far more nuanced than previously believed. A vital part of both innate and adaptive immune responses is played by AMPs, sometimes referred to as host defense peptides (HDPs). Numerous immunomodulatory processes are regulated by AMPs, including but not limited to (1) chemoattraction, (2) pro- and anti-inflammatory responses, cellular differentiation, (3) wound healing, and (4) enhanced bacterial death. Numerous immune cells, such as neutrophils, eosinophils, and macrophages, are chemoattracted to human cathelicidin LL-37.[22] Direct and indirect pathways are used by AMPs to attract immune cells. Increasing the expression of chemokines, signaling proteins that control chemotaxis in macrophages, mast cells, T cells, and epithelial cells, is one indirect example.[23,24] Hancock identified the signaling route through which LL-37 stimulates the synthesis of chemokines in keratinocytes, a kind of epithelial cell found in the top layer of the skin.[21] He showed that the Src family kinases (SFKs) are essential and that the P2X7 receptor,[25] SFKs, Akt (protein kinase B), and the transcription factors CREB and ATF1 are all part of the signaling pathway.
In the immunomodulation of both pro- and anti-inflammatory responses, AMPs also play significant roles. Human -defensins induce the expression of pro-inflammatory cytokines TNF- and IL-1 in human monocytes.[26] In vivo research by Falco and colleagues also revealed that the injection of HNP1 into rainbow trout (Onccorhynchus mykiss) caused the expression of genes encoding for pro-inflammatory cytokines like IL-1, TNF-, and IL-8, CC chemokines like CK5B, CK6, and CK7A, as well as genes involved in the synthesis of type I interferon (IFN).[27] -defensins, in contrast to -defensins, seem to have the opposite impact on inflammation. Human -Defensin-3 specifically inhibits inflammation that is NF-B (nuclear factor kappa-light chain-enhancer of activated B cells) dependent.
Mammalian and bacterial cells have highly diverse cell surface structures in terms of content and electrostatic charge (Figure 2). Drugs or drug delivery systems that can distinguish between the two have the potential to become antimicrobial therapeutics. Gram-negative bacteria like Escherichia coli and Pseudomonas aeruginosa have two lipid bilayer membranes that regulate the flow of materials and information: an outer membrane and an inner cellular (cytoplasmic) membrane. Due to the existence of porin, -barrel proteins that span the outside membrane and act as a pore, allowing molecules to passively permeate, the outer membrane is thought to be more "leaky".[28] The outer membrane's lipid content is arranged asymmetrically, with lipopolysaccharides (LPS) making up the majority of the outside leaflet and phospholipids making up the majority of the inner leaflet. LPS, commonly known as endotoxin, is a gram-negative bacteria-specific impurity that is frequently found in peptides and other biologics. A more symmetrical composition of phosphatidylethanolamine (70–80%), phosphatidylglycerol, and cardiolipin makes up the inner cytoplasmic membrane.

Figure 2. Comparison of the cell surface architectures of gram-negative and gram-positive bacteria.
Because bacterial and mammalian cell membranes have different chemical compositions, AMPs demonstrate cell selectivity. AMPs are promising therapeutic options due to their capacity to selectively destroy invading microorganisms without significantly damaging human host cells. Compared to mammalian cell membranes, bacterial cell membranes are typically more negatively charged. The positively charged AMP is drawn to the negatively charged bacterial cell surfaces, which in some situations cause the positively charged AMP to gather around the negatively charged surfaces. Both the structure of the membranes and the position of the anionic membrane molecules account for the variations and associated selectivity towards bacteria. Phospholipids, membrane proteins, steroids, and other substances make up the majority of membranes.[29] Acidic phospholipids such phosphatidylserine (PS), phosphatidylglycerol (PG), and cardiolipin (CL) are widely present in bacterial cell membranes (Figure 3). When found in the outer "leaflet" of the membrane bilayer, these phospholipids' head groups, which are negatively charged at physiological pH levels, might draw in positively charged AMPs. A thick layer of peptidoglycan, or the cell wall, is present in gram-positive bacteria, and this layer is highly functionalized with anionic glycopolymers known as wall teichoic acids (WTAs), which can protrude beyond the cell's outer membrane.[30] WTAs are absent, and peptidoglycans exclusively cover the surface of gram-negative bacteria. Instead, the asymmetric distribution of LPS in the outer leaflet of the outer membrane causes gram-negative bacteria to acquire more negative charge on their surface.
Mammalian cytoplasmic membranes are abundant in phospholipids such phosphatidylcholine (PC), phosphatidylethanolamine (PE), and sphingomyelin (SM), all of which are zwitterionic in normal settings, rendering the membrane generally neutral in charge. This is in contrast to bacterial cellular membranes. Zwitterionic phospholipids as well as phospholipids with negatively charged head groups can be found in mammalian cell membranes. The negatively charged phospholipids are distributed asymmetrically to the inner leaflet towards the cell interior, in contrast to bacterial membranes.[31] For instance, 98% of the total PE composition is found in the inner leaflet of the cellular membrane in bovine erythrocytes.[32]

Figure 3a. Comparison of membrane molecules in mammalians. Mammalian cell membranes contain zwitterionic phospholipids PC, PE, and neutral cholesterol.
Mammalian cell membranes are abundant in hydrophobic cholesterol and ergosterol molecules in addition to location differences in phospholipids.[33] Only hydrophobic interactions with AMPs are possible due to the relative neutrality and hydrophobicity of mammalian cell membranes, which are significantly weaker than the electrostatic contacts. Increased transmembrane potentials can also intensify these electrostatic interactions. Mammalian membrane potentials are roughly -100 mV, but bacterial transmembrane potentials are in the range of -140 mV. The selection of cells among AMPs may also be influenced by this greater negative membrane potential.[34]

Figure 3b. Comparison of membrane molecules in bacteria. Bacterial cell membranes have a higher abundance of anionic phospholipids PG, Lysyl PG, LPS, anionic polymer teichoic acid and organic molecule cardiolipin.
Similar pore-forming and membrane-thinning mechanisms seen in AMP-bacteria interactions are used by AMP's that target the membranes of cancer cells (i.e., membranolytic ACPs) to destabilize the membrane. This method of action needs to be separated from those peptides that target particular membrane-bound receptors, such as those targeted by RGD-containing peptide sequences in cancer cells. There are other intracellular targets for ACPs besides cellular membranes, such as mitochondrial plasma membranes. Gomesin, a cationic AMP discovered by Rodrigues and colleagues in 2008 from the Acanthoscurria gomesiana spider[40], has been demonstrated to have intracellular targets in melanoma cancer cells. Gomesin has a two stranded antiparallel -sheet structure. Targeting the endoplasmic reticulum (ER) membrane, gomesin induces cytotoxicity in melanoma cells well after translocation across the cellular membrane.[41] The ER membrane being lysed causes a large rise in cytosolic Ca2+ levels, overloading the mitochondria, and apoptosis.
For its therapeutic potential against cancer, LL-37 has been extensively investigated. Depending on the type of cancer and the tissue impacted, the interaction between cancer cells and LL-37 is complex and can have opposing effects. Tumorigenesis is reduced in some cancers, such as colon and gastric cancers, while it appears to be boosted in other cancers, such as ovarian, lung, and breast cancers. Apoptosis control by LL-37 may play a role in the etiology of malignant tumors, according to some studies.[42] Colon tumors and hematologic malignancies exhibit anti-tumor action as a result of LL-37-induced apoptosis in tumor cells.[43] Other tumor-suppressing mechanisms have been seen in the tissues of gastric tumors, including LL-37's capacity to block proteasomes and cause apoptosis by upsetting the balance of pro-growth cell cycle proteins.[44] However, LL-37 can also encourage the development of tumors in other tissues. For instance, in ovarian cancer tissue, LL-37 aids in the recruitment of mesenchymal stem cells (MSC) to increase the pathogenesis of malignant tumors and facilitate healing of the cancer cells.[45]
Although astounding at the time, the introduction of traditional antibiotics and subsequent overuse have led to bacterial pathogen strains that are resistant to antibiotics. More specifically, the global economic burden on health care costs has increased due to the emergence of multidrug-resistant (MDR) pathogens and the diseases they cause. There are two main ways that bacteria might become resistant to antibiotics: (1) gene mutation, or (2) acquiring genetic material by horizontal gene transfer (HGT). In the case of gene mutation, the mechanism of resistance takes the form of either a decrease in antibiotic absorption, a decrease in affinity for the substance, active ejection (also known as efflux) mechanisms, or adjustments to the cellular metabolism that lessen the antibiotic agent's adverse effects.
In both gram-negative and gram-positive bacteria, the chemical alteration and transformation of the antibiotic into a less harmful or innocuous form is one of the effective pathways to resistance. The creation of enzymes that may acetylate, phosphorylate, or adenylate an antibiotic to alter its hydrophobicity or other chemical properties has been documented. For instance, AMEs, or aminoglycoside modifying enzymes, can react with hydroxyl or amino groups. Some AMEs have developed into bifunctional enzymes that have phosphotransferase and acetylation activity.[46] The majority of the gentamicin- and methicillin-resistant strains of enterococci and S. aureus are caused by the advent of bifunctional AMEs.
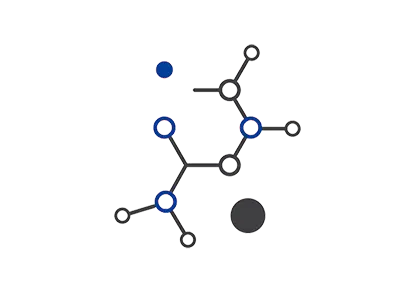
By dissolving the lactam ring in -lactam antibiotics like penicillins, cephalosporins, and cephamycins, -lactamases give bacteria another method of resistance (Figure 4). The antibiotic molecule is effectively "destroyed" by -lactamases when they hydrolyze the lactam ring and then proceed to decarboxylate it. -Lactamases were discovered in the 1940s, not long after penicillin started to be used extensively. The first -lactamase to be discovered was penicillinase, which Abraham and Chain isolated from Gram-negative E. coli in 1940.[47] Meticillin (also known as methicillin), a penicillin-class narrow-spectrum -lactam antibiotic, was created by Beecham in 1959 to fight the emergence of penicillin-resistant bacteria brought on by the existence of -lactamases.[48] The ortho-dimethoxyphenyl substituent adds bulk to the antibiotic, preventing -lactamases from binding to it and cleaving it. Meticillin directly inhibits the transpeptidase enzyme (i.e., penicillin-binding proteins (PBPs)), in a manner similar to other -lactam antibiotics that aim to impede the bacterial cell wall production by blocking cross linking of peptidoglycan polymers. PBPs are in charge of peptidoglycan polymers and D-alanyl-alanine peptide cross-linking. Metcillin was sarcastically referred to as a penicillinase-resistant -lactam antibiotic, despite the fact that it is no longer in use and has been superseded by newer analogs like cloxacillin.
It is not surprising that some AMPs have the capacity to control various types of cancer given the complicated immunomodulatory actions of AMPs. Anti-cancer peptides (ACPs), also known as AMPs that promote cytotoxicity on cancer cells, have been classified by mechanism as either membranolytic or non-membranolytic.[35] Although not all AMPs are ACPs, negatively charged membranes are a feature of both cancer and bacterial cells. Cancer cells have a negatively charged surface that is reminiscent of bacterial cell membranes because they contain anionic membrane components such phosphatidylserines (PS), glycosaminoglycans, heparan sulfate, O-glycosylated mucins, sialylated gangliosides, and glycoproteins. AMPs that are ACPs can selectively target cancer over healthy mammalian cells, much like they can with bacterial cells. Contrary to the typical membrane asymmetry observed in normal cells, cancer cells collect PS during cell transformation on the outside (presenting) leaflet of the cell membrane. Additionally, neutral cholesterol molecules are more plentiful in normal cells, increasing membrane stability and AMP resistance. The majority of studies concur that compared to normal cell membranes, cancer cell membranes are more fluid.[36-38] However, cancer cells, particularly those located in prostate and breast tissues, can occasionally have increased levels of cholesterol in their membranes as a means of dodging AMPs, similar to the defenses put forth by bacteria.[39]
Because AMPs have several and varied mechanisms of action on bacteria, they are less likely to encourage the kind of resistance found in bacteria with conventional antibiotics. The majority of conventional antibiotics have a very specific mode of action that is frequently stopped by a single gene mutation. The cell membrane and several intracellular targets that induce death are only a few of the areas of the bacteria that are targeted by AMPs. Due to their millions of years of coevolution with bacteria, AMPs have had the opportunity to develop sequences that are both effective antibiotics and resistant to widespread forms of resistance. Although some AMP responses have been documented, resistance to these drugs is less widespread and targeted than it is to conventional antibiotics. A gram-positive bacterium called S. aureus, for instance, has developed a universal mechanism to lower the charge on its surface by integrating D-alanine into the polymers of teichoic acid.[49] A L-lysine residue can also be transferred enzymatically from S. aureus to the membrane lipid PG to create lysyl-PG.[50] As a result of this procedure, PG has a net charge that ranges from -1 to +1, which lessens electrostatic interactions with cationic AMPs.

Figure 4. Lactam antibiotics penicillins, cephalosporins, and cephamycins. The R groups (R, R1, R2) are variable with full structures not shown for clarity. The red color denotes the 4-membered lactam ring in each derivative (top row). The mechanism of β-lactamase action on penicillin showing two steps starting with a ring-opening hydrolysis of the lactam followed by decarboxylation (bottom row).
Despite valiant attempts over the past ten years, the US-FDA has not yet approved any systemically dosed peptide antibiotics. This failure is partly attributable to expensive peptide production and low market demand for some medicinal AMPs. The 22-amino-acid cationic magainin 2 derivative Pexiganan was the first antimicrobial peptide to enter clinical development. Magainin was discovered by the Zasloff group in 1987 and was initially isolated from the skin of the African clawed frog (Xenopus laevis).[51] Pexiganan performed remarkably well in vitro during the preclinical stage against a wide range of microorganisms. As an alternative to the existing systemic dosed antibiotics, pexiganan was initially developed as a topical locally applied antibiotic treatment. The peptide is vulnerable to proteolytic breakdown in the blood stream, which lowers its bioavailability; however, local applications at the infection site can result in significantly higher quantities. In comparison to the anticipated 1.0 gram of intravenous dose needed for conventional antibiotics, substantially less peptide is needed for topical treatment. Pexiganan was first developed at a cost of $1000/g, making cGMP manufacture unaffordable for a medication that did not considerably outperform existing small-molecule antibiotics. However, the FDA suspended approval as Pexiganan's development entered phase III of the clinical trials because of erratic findings and worries about the manufacturing process.[52] Depexium Pharmaceuticals has recently finished phase III of the development of pexiganan, which is being used as a topical cream to treat diabetic foot infections.
Plectasin was initially isolated from the saprophytic ascomycete Pseudoplectania nigrella, a novel antibacterial defensin peptide that was identified by Mygind in 2005.[53] Dr. Hans-Henrik Kristensen oversaw the research and clinical development of Plectasin at Novozymes. They observed that the defensin peptide (plectasin) excreted by the fungus shared a high degree of homology with defensins found in insects and mussels in terms of secondary structures. The antibacterial compound plectasin has a restricted antibacterial spectrum and worked best against Streptococcus pneumonia. Intravenously given peptide was equivalent to or superior to traditional antibiotic therapy, according to in vivo investigations. Defensin-like peptides are extremely resistant to being broken down by proteolytic enzymes because of their compact and restricted structure, which is partly attributable to the three disulfide bridges. Because of this, Plectasin can be completely intact recovered from the urine of treated animals. This peptide appeared to be the perfect remedy for penicillin-resistant Pneumococcus because it is a limited spectrum antibiotic. The decision was reached to stop the development of the peptide in its current form because to the tiny market size of patients with Pneumococcus infections as well as the high expense of clinical development and commercial manufacture. With a novel analogue called NZ2114 that shown systemic efficacy against S. aureus, further efforts were made to increase the antibacterial activity of Plectasin. 2010 saw the signing of a contract between Novozymes and Sanofi-Aventis for the clinical development of NZ2114.
While AMP clinical testing is still ongoing, there are still several difficulties and obstacles to FDA approval. Although producing cGMP peptides is expensive, costs are starting to decline as a result of improved chemistries that increase yields and lower starting material costs. For instance, over the past 20 years, protected amino acids have become significantly more affordable. Only lately have peptides that are entirely composed of protected D-forms of amino acids,[54] designed for increased protease resistance, entered clinical research. This is partly because protected D-forms of amino acids are now more affordable. When developing medications for intravenous administration, peptide stability in general and their capacity to withstand proteolytic breakdown are crucial considerations. The sequence is made less accessible to enzymes by the presence of D-amino acids as well as highly restricted peptides like defensins that make use of numerous disulfide bridges. The addition of hydrocarbon-staples is another technique for preserving peptide stability, particularly for secondary structures like alpha-helices.[55-57] Compared to the native sequence, peptide stapling can produce stronger proteolytic degradation resistance; nevertheless, it does not always result in higher antibacterial efficacy. Additional bespoke peptide modifications, such as macrocyclization,[59-60] that tighten peptide backbone conformations can be added. (for instance, sidechain-to-sidechain, head-to-tail, etc.), oxidation,[61–62] Click chemistry, N-alkylation, and other terms to learn more.
_______________________________________________________________
References
Zeya, H. I., and J. K. Spitznagel. Journal of Bacteriology 91.2 (1966): 755-762.
Zeya, H. I., and J. K. Spitznagel. Journal of Bacteriology 91.2 (1966): 750-754.
Zeya, H. I., and J. K. Spitznagel. Science (1966): 1049-1051.
Zeya, H. I., J. K. Spitznagel, and J. H. Schwab. Proceedings of the Society for Experimental Biology and Medicine 121.1 (1966): 250-253.
European Centre for Disease Prevention and Control Antimicrobial Resistance Interactive Database (EARS-NET) data for 2013.
Yotova Maya, et al. International Journal of Nutrition and Food Sciences. Vol. 5, No. 6-1, 2016
Dathe, Margitta, and Torsten Wieprecht. Biochimica et Biophysica Acta (BBA)-Biomembranes 1462.1 (1999): 71-87.
Selsted, Michael E., et al. Journal of Biological Chemistry 268.9 (1993): 6641-6648.
Bulet, Phillipe, et al. Developmental & Comparative Immunology 23.4 (1999): 329-344.
Shai, Yechiel. Peptide Science 66.4 (2002): 236-248.
Hancock, Robert EW. The Lancet Infectious Diseases 1.3 (2001): 156-164.
Risso, Angela. Journal of Leukocyte Biology 68.6 (2000): 785-792.
Nicolas, Pierre. The FEBS Journal 276.22 (2009): 6483-6496.
Mansour, Sarah C., Olga M. Pena, and Robert EW Hancock. Trends in immunology 35.9 (2014): 443-450.
Yedery, R. D., and K. V. R. Reddy. The European Journal of Contraception & Reproductive Health Care 10.1 (2005): 32-42.
Srakaew, Nopparat, et al. Human Reproduction 29.4 (2014): 683-696.
Huang, C.C., Couch, G.S., Pettersen, E.F., and Ferrin, T.E. Pacific Symposium on Biocomputing 1:724 (1996).
Sitaram, Narasimhaiah, Chilukuri Subbalakshmi, and Ramakrishnan Nagaraj. Biochemical and Biophysical Research Communications 309.4 (2003): 879-884.
Puri, Sumant, and Mira Edgerton. Eukaryotic Cell 13.8 (2014): 958-964.
Lee, Juneyoung, et al. Free Radical Biology and Medicine 52.11 (2012): 2302-2311.
Jiao, Delong, et al. Scientific Reports 7 (2017).
Wan, Min, et al. Journal of Leukocyte Biology 95.6 (2014): 971-981.
Mookherjee, N., and R. E. W. Hancock. Cellular and Molecular Life Sciences 64.7-8 (2007): 922.
Nijnik, Anastasia, et al. Journal of Innate Immunity 4.4 (2012): 377-386.
Bartlett, Rachael, Leanne Stokes, and Ronald Sluyter. Pharmacological Reviews 66.3 (2014): 638-675.
Chaly, Yu V., et al. European Cytokine Network 11.2 (2000): 257-266.
Falco, A., et al. Fish & Shellfish Immunology 24.1 (2008): 102-112.
Koebnik, Ralf, Kaspar P. Locher, and Patrick Van Gelder. Molecular Microbiology 37.2 (2000): 239-253.
Jiang, Ziqing, et al. Chemical Biology & Drug Design (2017).
Brown, Stephanie, John P. Santa Maria Jr, and Suzanne Walker. Annual Review of Microbiology (2013): 67, 313-336.
Zasloff, Michael. Nature 415.6870 (2002): 389-395.
Takahashi, Daisuke, et al. Biochimie 92.9 (2010): 1236-1241.
Lai, Yuping, and Richard L. Gallo. Trends in Immunology 30.3 (2009): 131-141.
Yeaman, Michael R., and Nannette Y. Yount. Pharmacological Reviews 55.1 (2003): 27-55.
Gaspar, Diana, A. Salomé Veiga, and Miguel ARB Castanho. Frontiers in Microbiology 4 (2013).
Utsugi, Teruhiro, et al. ” Cancer Research 51.11 (1991): 3062-3066.
Hoskin, David W., and Ayyalusamy Ramamoorthy. Biochimica et Biophysica Acta (BBA)-Biomembranes 1778.2 (2008): 357-375.
Schweizer, Frank. European Journal of Pharmacology 625.1 (2009): 190-194.
Li, Ying Chun, et al. The American Journal of Pathology 168.4 (2006): 1107-1118.
Rodrigues, Elaine G., et al. Neoplasia 10.1 (2008): 61-68.
Paredes-Gamero, Edgar J., et al. Molecular Pharmaceutics 9.9 (2012): 2686-2697.
Ouyang L, Shi Z, Zhao S et al. Cell Prolif. (2012) 45:487–498
(a) Mader JS, Mookherjee N, Hancock RE et al. Mol Cancer Res (2009) 7:689–702. (b) Ren SX, Cheng AS, To KF et al. Cancer Res (2012) 72:6512–6523. (c) Ren SX, Shen J, Cheng AS et al. PLoS One. (2013) 8:e63641.
Wu WK, Sung JJ, To KF et al (2010) J Cell Physiol 223:178–186.
(a) Coffelt SB, Marini FC, Watson K et al. Proc Natl Acad Sci USA. (2009) 106:3806–3811. (b) Wu WK, Wang G, Coffelt SB et al (2010) J Int Cancer 127:1741–1747.
Hollenbeck, Brian L., and Louis B. Rice. Virulence 3.5 (2012): 421-569.
Abraham, Edward P., and Ernst Chain. Nature 146 (1940): 837.
Dickinson, JM, and NB Pride. Arch. Intern. Med 104 (1959): 180.
Peschel, Andreas, and L. Vincent Collins. Peptides 22.10 (2001): 1651-1659.
Andrä, Jörg, et al. Journal of Biological Chemistry 286.21 (2011): 18692-18700.
Zasloff M. Proceedings of the National Academy of Sciences of the United States of America 1987; 84(15):5449-5453.
Yotova Maya, et al. International Journal of Nutrition and Food Sciences Vol. 5, No. 6-1, 2016, pp. 1-4.
Mygind, Per H., et al. Nature 437.7061 (2005): 975.
de la Fuente-Núñez, César, et al. Chemistry & Biology 22.2 (2015): 196-205.
Chapuis, Hubert, et al. Amino Acids 43.5 (2012): 2047-2058.
Kutchukian, Peter S., et al. Journal of the American Chemical Society 131.13 (2009): 4622-4627.
Walensky, Loren D., and Gregory H. Bird. Journal of Medicinal Chemistry 57.15 (2014): 6275-6288.
www.cpcscientific.com
Young, Travis S., et al. Proceedings of the National Academy of Sciences 108.27 (2011): 11052-11056.
Leone, Marilisa, et al. Chemical Biology & Drug Design 77.1 (2011): 12-19.
Gounder, Anshu P., et al. Journal of Biological Chemistry 287.29 (2012): 24554-24562.
Wiens, Mayim E., and Jason G. Smith. Journal of Virology 89.5 (2015): 2866-2874.
Zhu, S., et al. Journal of Thrombosis and Haemostasis (2016), 14: 1070–81.
Crizer, David M., and Scott A. McLuckey. Journal of the American Society for Mass Spectrometry 20.7 (2009): 1349-1354.

Please submit a detailed description of your request. We will provide you with professional services to meet your research requests. You can also send emails directly to sales@chinesepeptide.com for inquiries.

